Aug. 24 2017 @ 石川県立大
この資料のURL ⇒ http://ynlab.info/training201708/
第一部
I. DDBJ・遺伝研スパコン入門(プレゼン)
1708DDBJ導入.pdf (6.9MB) – プレゼンPDF (8/23 update 版)
II. DDBJパイプライン紹介(プレゼン・guest体験)
1708パイプライン入門.pdf (9.2MB) – プレゼン PDF (8/23 update 版)
mapped.zip (891MB) – DDBJ pipeline で SRA044892 を TAIR10 ゲノムにマッピングした結果 (out2.bam, out2.bam.bai) 詰め合わせ。ダウンロードして iGV でながめてみましょう。
【tips】
iGV で以下のようなエラーが出る場合:

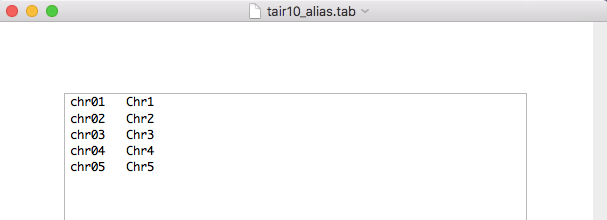
以下のような染色体名の別名ファイルを作って(空白は TAB です)ユーザフォルダの直下にある igv フォルダに置きましょう(この場合は TAIR10の 染色体別名)
第二部
I. DDBJ・遺伝研スパコンのハードとソフト超入門
1708スパコン入門.pdf (2.7MB) – プレゼン PDF (8/23 update 版)
※ 今回の講習では、ここまで(スパコンにログインするまで)をひとまずの到達点とします。以下はログインできるようになったら自習してみてください。すでにログインできるようになっている方は、時間内に以下の手順を先にすすめていただいて構いません!質問はご自由に。
【こちらもおすすめ!】コマンドライン講習会(東大情報生命・笠原さんの教材)
II. Unix 基本コマンドの超入門
1. 基本コマンドの練習をしましょう
リダイレクト。ディレクトリ表示
- > : プログラムから書き込み
- < : プログラムへ書き込み
- | : プログラムからプログラムに出力を渡す
- . : 自分がいるディレクトリを示す
- .. : 自分がいる一つ上の階層のディレクトリを示す
- ~ : 自分のホームディレクトリを示す
ディレクトリ表示(一番使うコマンド)
- ls : (LiSt) ファイル名をリストする
ファイル閲覧関係
- cat : (CATenate) ファイルの連結・表示
- head : ファイルの先頭を表示
- less : ファイルをページ単位で表示 [スペースでページ送り、Bでページ戻り、Q で終了]
$ ls
ddbj_database training.tar lcl
$ ls -l # -l オプションをつけると表示される情報が変わる
合計 74744
drwxr-xr-x 3 tafujisa yn-nig 4096 12月 27 11:48 2012 ddbj_database
-rw-r--r-- 1 yanakamu yn-nig 76523520 12月 15 17:48 2016 training.tar
drwxr-xr-x 11 yanakamu yn-nig 4096 12月 15 13:35 2016 lcl
$ ls -a # -a オプションをつけると不可視ファイル (. で始まるファイル、ディレクトリが見える)
. .bash_profile .emacs.d .login .pyenv .viminfo
.. .bashrc .gem .matplotlib .rbenv .zprofile
.RepeatMaskerCache .cache .gnome2 .mc .screenrc .zshrc
.Xauthority .ddbjing34 .gnuplot_history .mozilla .ssh ddbj_database
.aspera .ddbjing34_deleteme .history .mysql_history .subversion training.tar
.bash_history .emacs .lesshst .pki .tcshrc lcl
ls の出力からファイルをつくってみます。cat で閲覧します。
$ ls > test # ls の出力をファイル test をつくって流し込む
$ ls
ddbj_database training.tar lcl test
$ cat test # ファイル test の中身を表示する(画面上と違い、一行一ファイルになります)
ddbj_database
training.tar
lcl
testファイル&ディレクトリ(フォルダ)操作関係
- cp : (CoPy) ファイルのコピー
- mv : (MoVe) ファイルの移動、名称変更
- rm : (ReMove) ファイルの削除
$ ls
ddbj_database training.tar lcl test
$ cp test test2 # ファイル test を test2 にコピーする
$ cat test2 # test2 を表示する
ddbj_database
training.tar
lcl
test
$ cat test test2 > test3 # test と test2 の中身を合わせて test3に書き出す
$ cat test3 # test3 を表示する
ddbj_database
training.tar
lcl
test
ddbj_database
training.tar
lcl
test
$ mv test3 test4 # test3 を test4 に名称変更する
$ ls
ddbj_database training.tar lcl test test2 test4
$ rm test4 # test4 を消す。黙って消えるので注意
$ ls
ddbj_database training.tar lcl test test2ディレクトリ移動、作成関係
- mkdir / rmdir : (Make Directory / ReMove Directory) ディレクトリの作成・削除
- pwd : (Print Working Directory) 今いる場所を表示する
- cd : (Change Directory) いる場所を変更する
$ mkdir testdir # testdir というディレクトリ(フォルダ)を作る
$ ls
ddbj_database training.tar lcl test test2 testdir
$ cd testdir # testdir に入る
$ pwd # 今いるディレクトリを表示する(yanakamu の部分はあなたのユーザ名になります)
/home/yanakamu/testdir
$ ls
$ mv ../test ./ # 一つ下のディレクトリ (..) から test ファイルをここ (.) に移動する
$ ls
test
$ cd .. # 一つ下のディレクトリに移動する
$ pwd # 今いるディレクトリを表示する(yanakamu の部分はあなたのユーザ名になります)
/home/yanakamu
$ ls # test が testdir に移動したのでここからは消えている
ddbj_database training.tar lcl test2 testdir
$ rmdir testdir # testdir を消そうとするが、空ではないので消えない
rmdir: failed to remove `testdir': ディレクトリは空ではありません
$ rm testdir/test # testdir の中の test ファイルを消す。黙って消える
$ ls testdir # testdir の中を表示する、空になっている
$ rmdir testdir # 今度は testdir を消すことができる
$ ls
ddbj_database training.tar lcl test2アーカイブ関係
- tar : (Tape ARchive) アーカイブの操作(ファイルの取りまとめ)
- zip / unzip : ファイル圧縮・展開
- gzip / gunzip : GNU ファイル圧縮・展開
$ cp test2 test3
$ ls
ddbj_database training.tar lcl test2 test3
$ tar -cf test.tar test2 test3 # test.tar という名前で test2 と test3 を束ねる
$ ls
ddbj_database training.tar lcl test.tar test2 test3
$ tar -tf test.tar
test2
test3
$ rm test2 test3 # test2 test3 を消しちゃう
$ ls
ddbj_database training.tar lcl test.tar
$ ls -l
合計 74756
drwxr-xr-x 3 tafujisa yn-nig 4096 12月 27 11:48 2012 ddbj_database
-rw-r--r-- 1 yanakamu yn-nig 76523520 12月 15 17:48 2016 training.tar
drwxr-xr-x 11 yanakamu yn-nig 4096 12月 15 13:35 2016 lcl
-rw-r--r-- 1 yanakamu yn-nig 10240 12月 16 13:48 2016 test.tar
$ gzip test.tar # test.tar を圧縮する
$ ls -l
合計 74744
drwxr-xr-x 3 tafujisa yn-nig 4096 12月 27 11:48 2012 ddbj_database
-rw-r--r-- 1 yanakamu yn-nig 76523520 12月 15 17:48 2016 training.tar
drwxr-xr-x 11 yanakamu yn-nig 4096 12月 15 13:35 2016 lcl
-rw-r--r-- 1 yanakamu yn-nig 192 12月 16 13:48 2016 test.tar.gz
$ gunzip test.tar.gz
$ ls
ddbj_database training.tar lcl test.tar
$ tar -xf test.tar # test.tar の中身 (test2, test3 を取り出す)
$ ls
ddbj_database training.tar lcl test.tar test2 test3
$ cat test2 test3
ddbj_database
training.tar
lcl
test
ddbj_database
training.tar
lcl
test
$ rm test.tar test2 test3 # 今作業したファイルをカタ付ける
$ ls
ddbj_database training.tar lcl # 最初からあったファイルだけが表示されたら ok ですII. マッピング演習
tophat2 を使って、真核生物(酵母)のゲノムにRNA-seqのNGSリードをマッピングし、それを多くのリードセットに適用するためのスクリプトを使ってみましょう。スクリプトを作ってみましょう、と言いたいところですが、Unix上で文書・スクリプトを編集するための「エディタ」の使い方を覚えるのがちょっとハードルが高いので、事前にスクリプトを用意してあります。
代表的なマッピングツールの3種の特徴をめっさざっくりと紹介しますと:
- Bowtie: ギャップを許容しない
- BWA: ギャップを許容する(スプライシングを考慮するわけではない)
- TopHat: リードが 2 つのエクソンにまたがっている場合も対応できる。つまりスプライシングを考慮してしてくれるので便利。そのかわり処理に時間がかかる。RNA-seq をマッピングするのに使うことが多い。
1. スクリプト・データセットをダウンロードしましょう。
$ curl http://ynlab.info/data/training.tar.gz > training.tar.gz手打ちするのは面倒ですね?間違えやすいし。
上記の画面からターミナル画面にコピー&ペーストすればokです。おっと、先頭の $ はそれがシェルのコマンドであることを示している記号で、コマンドの一部ではありません。コピーしないでくださいね。
2. スクリプト・データセットの圧縮を解いて展開しましょう
$ tar -xzf training.tar.gz
$ ls
training training.tar
$ cd training
$ ls
Annotation Sequence fastq mkindex.sh tophat.sh
cd Annotation
$ ls
genes.gtf
$ cd ..
$ cd Sequence
$ ls
Bowtie2Index
$ cd Bowtie2Index
$ ls
genome.1.bt2 genome.3.bt2 genome.fa genome.rev.2.bt2
genome.2.bt2 genome.4.bt2 genome.rev.1.bt2
$ cd ../..
$ ls
Annotation Sequence fastq mkindex.sh tophat.sh
$ cd fastq
$ ls
SRR453567_R1.fastq SRR453568_R1.fastq SRR453569_R1.fastq
SRR453570_R1.fastq SRR453571_R1.fastq SRR453567_R2.fastq
SRR453568_R2.fastq SRR453569_R2.fastq SRR453570_R2.fastq
SRR453571_R2.fastq
$ cd ..
$ pwd
/home/yanakamu/training
$ tophat2
-bash: tophat2: コマンドが見つかりませんおや、スパコンに tophat2 がないぞ?どういうこと?
こまったらググれ(sc.ddbj.nig.ac.jp サイト内で、tophat2 をキーワードに調べる方法)
google 検索窓で [ site:sc.ddbj.nig.ac.jp tophat2 ]
https://sc.ddbj.nig.ac.jp/index.php/ja-avail-oss ← ここに書いてあるよ
用意したデータ
元論文: Nookaew, I et al., (2012) A comprehensive comparison of RNA-Seq-based transcriptome analysis from reads to differential gene expression and cross-comparison with microarrays: a case study in Saccharomyces cerevisiae. Nucl. Acids Res., 40: 10084-10097. doi: 10.1093/nar/gks804
リファレンス: S. cerevisiae Ensembl R64-1-1 / iGenomes から取得
NGSリード: SRA051410 ( RUN’s SRR453567, SRR453568, SRR453569, SRR453570, SRR453571 ) / DRAsearch から取得
3. tophat2の実行の仕方
1 run 分だけ手で実行してみましょう。
$ $ /usr/local/pkg/tophat2/current/tophat2 -p 2 -o SRR453567 -G ./Annotation/genes.gtf ./Sequence/Bowtie2Index/genome ./fastq/SRR453567_R1.fastq ./fastq/SRR453567_R2.fastq
[2017-08-22 16:25:21] Beginning TopHat run (v2.1.1)
-----------------------------------------------
[2017-08-22 16:25:21] Checking for Bowtie
Bowtie version: 2.3.2.0
[2017-08-22 16:25:21] Checking for Bowtie index files (genome)..
[2017-08-22 16:25:21] Checking for reference FASTA file
[2017-08-22 16:25:21] Generating SAM header for ./Sequence/Bowtie2Index/genome
[2017-08-22 16:25:21] Reading known junctions from GTF file
[2017-08-22 16:25:21] Preparing reads
left reads: min. length=101, max. length=101, 9997 kept reads (3 discarded)
right reads: min. length=101, max. length=101, 9992 kept reads (8 discarded)
[2017-08-22 16:25:22] Building transcriptome data files SRR453567/tmp/genes
[2017-08-22 16:25:23] Building Bowtie index from genes.fa
[2017-08-22 16:25:30] Mapping left_kept_reads to transcriptome genes with Bowtie2
[2017-08-22 16:25:31] Mapping right_kept_reads to transcriptome genes with Bowtie2
[2017-08-22 16:25:32] Resuming TopHat pipeline with unmapped reads
[2017-08-22 16:25:32] Mapping left_kept_reads.m2g_um to genome genome with Bowtie2
[2017-08-22 16:25:32] Mapping left_kept_reads.m2g_um_seg1 to genome genome with Bowtie2 (1/4)
[2017-08-22 16:25:32] Mapping left_kept_reads.m2g_um_seg2 to genome genome with Bowtie2 (2/4)
[2017-08-22 16:25:33] Mapping left_kept_reads.m2g_um_seg3 to genome genome with Bowtie2 (3/4)
[2017-08-22 16:25:33] Mapping left_kept_reads.m2g_um_seg4 to genome genome with Bowtie2 (4/4)
[2017-08-22 16:25:33] Mapping right_kept_reads.m2g_um to genome genome with Bowtie2
[2017-08-22 16:25:34] Mapping right_kept_reads.m2g_um_seg1 to genome genome with Bowtie2 (1/4)
[2017-08-22 16:25:34] Mapping right_kept_reads.m2g_um_seg2 to genome genome with Bowtie2 (2/4)
[2017-08-22 16:25:34] Mapping right_kept_reads.m2g_um_seg3 to genome genome with Bowtie2 (3/4)
[2017-08-22 16:25:34] Mapping right_kept_reads.m2g_um_seg4 to genome genome with Bowtie2 (4/4)
[2017-08-22 16:25:34] Searching for junctions via segment mapping
[2017-08-22 16:25:35] Retrieving sequences for splices
[2017-08-22 16:25:35] Indexing splices
Building a SMALL index
[2017-08-22 16:25:36] Mapping left_kept_reads.m2g_um_seg1 to genome segment_juncs with Bowtie2 (1/4)
[2017-08-22 16:25:36] Mapping left_kept_reads.m2g_um_seg2 to genome segment_juncs with Bowtie2 (2/4)
[2017-08-22 16:25:36] Mapping left_kept_reads.m2g_um_seg3 to genome segment_juncs with Bowtie2 (3/4)
[2017-08-22 16:25:36] Mapping left_kept_reads.m2g_um_seg4 to genome segment_juncs with Bowtie2 (4/4)
[2017-08-22 16:25:36] Joining segment hits
[2017-08-22 16:25:37] Mapping right_kept_reads.m2g_um_seg1 to genome segment_juncs with Bowtie2 (1/4)
[2017-08-22 16:25:37] Mapping right_kept_reads.m2g_um_seg2 to genome segment_juncs with Bowtie2 (2/4)
[2017-08-22 16:25:37] Mapping right_kept_reads.m2g_um_seg3 to genome segment_juncs with Bowtie2 (3/4)
[2017-08-22 16:25:37] Mapping right_kept_reads.m2g_um_seg4 to genome segment_juncs with Bowtie2 (4/4)
[2017-08-22 16:25:37] Joining segment hits
[2017-08-22 16:25:37] Reporting output tracks
-----------------------------------------------
[2017-08-22 16:25:40] A summary of the alignment counts can be found in SRR453567/align_summary.txt
[2017-08-22 16:25:40] Run complete: 00:00:19 elapsed
おわりました。結果をちら見してみましょう。
$ ls
Annotation/ SRR453567/ Sequence/ fastq/ mkindex.sh* tophat.sh*
$ cd SRR453567
$ ls
accepted_hits.bam align_summary.txt insertions.bed logs/ unmapped.bam
accepted_hits.bam.bai deletions.bed junctions.bed prep_reads.info
$ cat align_summary.txt
Left reads:
Input : 10000
Mapped : 8354 (83.5% of input)
of these: 546 ( 6.5%) have multiple alignments (48 have >20)
Right reads:
Input : 10000
Mapped : 7271 (72.7% of input)
of these: 462 ( 6.4%) have multiple alignments (43 have >20)
78.1% overall read mapping rate.
Aligned pairs: 6739
of these: 433 ( 6.4%) have multiple alignments
32 ( 0.5%) are discordant alignments
67.1% concordant pair alignment rate.
たくさんあるので自動化したい⇒「スクリプト」を書いて、qsub で queue に入れます。
4. スクリプト化
$ cd ..
$ pwd
/home/yanakamu/training
$ ls
Annotation SRR453567 Sequence fastq mkindex.sh tophat.sh
$ cat tophat.sh
#!/bin/bash
#$ -S /bin/bash
#$ -pe def_slot 2
#$ -cwd
f=("SRR453567" "SRR453568" "SRR453569" "SRR453570" "SRR453571")
for file in ${f[@]}
do
/usr/local/pkg/tophat2/current/tophat2 -p 2 -o ${file} -G ./Annotation/genes.gtf \
./Sequence/Bowtie2Index/genome ./fastq/${file}_R1.fastq ./fastq/${file}_R2.fastq
done
#end
これは bash のスクリプトです。画面上で編集するには、Emacs, vim, nano editor などを使います。操作を覚えるのが結構大変なので、最初のうちは Mac/PC 上でつくって送信すると良いかもしれません。
qsub で queue に入れます。qstat で実行状況をみる。qdel でジョブを削除するなどができます。
参照:スパコン・基本的利用方法
$ qsub -l short tophat.sh
Your job 36553768 ("tophat.sh") has been submitted
$ qstat
job-ID prior name user state submit/start at queue jclass slots ja-task-ID
------------------------------------------------------------------------------------------------------------------------------------------------
36550909 0.25032 QLOGIN yanakamu r 12/16/2016 09:18:09 login.q@t266i 1
36550910 0.25032 QLOGIN yanakamu r 12/16/2016 09:18:15 login.q@t266i 1
36553768 0.25013 tophat.sh yanakamu r 12/16/2016 11:41:43 short.q@t122i 2
36550172 0.25254 tophat.sh yanakamu qw 12/15/2016 17:49:08 2
$ qdel 36550172
yanakamu has deleted job 36550172
$ qstat
job-ID prior name user state submit/start at queue jclass slots ja-task-ID
------------------------------------------------------------------------------------------------------------------------------------------------
36550909 0.25034 QLOGIN yanakamu r 12/16/2016 09:18:09 login.q@t266i 1
36550910 0.25034 QLOGIN yanakamu r 12/16/2016 09:18:15 login.q@t266i 1
$ ls
Annotation SRR453569 Sequence tophat.sh tophat.sh.pe36553768
SRR453567 SRR453570 fastq tophat.sh.e36553768 tophat.sh.po36553768
SRR453568 SRR453571 mkindex.sh tophat.sh.o365537685. IGVで可視化する
IGV はスパコン上で動作させるのではなく、アウトプットをダウンロードしてきて、手元のMac/PC上で閲覧します。
The Integrative Genomics Viewer (IGV) はBROAD Instituteから取得できます(ググれ)
http://software.broadinstitute.org/software/igv/
まず、スパコン上でtophat のアウトプットのbamファイルからインデックスファイルをつくります。
$ ls
Annotation SRR453567 Sequence fastq mkindex.sh tophat.sh
$ cd SRR453567
$ ls
accepted_hits.bam deletions.bed junctions.bed prep_reads.info
align_summary.txt insertions.bed logs unmapped.bam
$ samtools index accepted_hits.bam
$ ls
accepted_hits.bam align_summary.txt insertions.bed logs unmapped.bam
accepted_hits.bam.bai deletions.bed junctions.bed prep_reads.infoaccepted_hits.bai ができている。
accepted_hits.bam と accepted_hits.bam.bai の両方をデスクトップに持ってくる。
*これはスパコンではなく Mac の画面です*
yn@ynAir:~ $ scp "yanakamu@gw.ddbj.nig.ac.jp:~/training/SRR453567/accepted_hits.bam*" .
accepted_hits.bam 100% 1685KB 1.7MB/s 00:00
accepted_hits.bam.bai 100% 18KB 17.7KB/s 00:00
yn@ynAir:~ $ ls
Desktop Library accepted_hits.bam Documents Movies accepted_hits.bam.baiCyberduck (Mac) や WinSCP (Win) を使うと、ドラッグ&ドロップでファイルの転送ができるので、入手をおすすめします。
IGV からメニュー File>Load from File… で accepted_hits.bam を読み込む ⇒ 表示される。
【関連解析例】
- クオリティの低いリードの除去を行う
- cufflinks で FPKM (Fragments Per Kilobase of exon per Million mapped fragments)を調べる
- cuffdiff でサンプル間の FPKM を比較する
- RのcummeRbundを使って解析、作図する
どうやったらできるようになる?
- 論文を読んでがんばる(そう詳しくは書いてない)
- 書籍を参照する
- google 検索⇒サイト・動画を参照する
などの方法があります。講習の機会に DDBJ や DBCLS の人々に聞くのも良いでしょう。
おすすめ教材
- 書籍「次世代シークエンサーDRY解析教本-細胞工学別冊-」(Macで全部できる)
- 動画 平成28年度 次世代シークエンサ(NGS)ハンズオン講習会(バイオインフォマティクス人材育成カリキュラム)
- サイト (Rで)塩基配列解析(東大・門田さん)スゴイ情報量
- 書籍「トランスクリプトーム解析」(上記のサイトの書籍版)
他にもいろいろありますよ。適切なキーワードでうまく検索するスキルを身に着けよう。
謝辞
- 参照ゲノム、RNA-seqリードの選択、スクリプトは2014年ゲノム支援講習会の千葉大学・高橋さんの講習内容を踏襲させていただきました。
- 説明の流れは「Dry解析教本」岩手医科大学・大桃さんの章を参考にさせていただいてきました。
- 本文中ターミナルのイメージを表示するのに、WordPress プラグイン「WP Code Highlight.js」を利用しています。